4/20/2023:
Infectious bronchitis virus (IBV) is the Gammacoronavirus responsible for infectious bronchitis in chicken. Like its distant cousin SARS-CoV-2, IBV is subject to the rapid evolutionary change that is characteristic of RNA viruses. As a result of this, IBV is a remarkably diverse pathogen harbouring dozens of lineages that are in competition for infection of new hosts. Study of the genetic sequences of IBV has provided several interesting insights into the churning dynamics of this important pathogen.
As the largest veterinary laboratory in the Netherlands, Royal GD has acquired a sizeable database of genetic sequences derived from clinical samples which were submitted to our laboratory. These concern sequences of a fragment of the S gene which covers one of three hypervariable regions (HVR) of the Spike protein. Highly variable sequences are attractive regions for sequence analysis since these provide high resolution of virus variants. In addition, these HVRs have been shown to encode parts of the spike protein that are targeted by the avian immune system. In order to learn more about the dynamics of IBV lineages, this sequence collection was subjected to an investigation. The analysis was restricted to Dutch submissions during a ten-year period. The detailed results of this investigation were recently published.
Besides a handful of minor variants, two major IBV lineages co-exist in the Netherlands. One major lineage is GI-13, also known as 793B and the other is GI-19 often referred to as QX. Co-existence of multiple viral lineages is not unique and is known for other viruses. Often in such cases, a pair of major lineages is engaged in epidemic switching, in which the two lineages alternate as the dominant variant. However, in IBV this did not appear to be the case. Instead of regular switching, the relative frequencies of the two lineages were stable throughout the investigated period. So what could explain this peaceful co-existence?
Some clue may be found when considering the production type of the host. We found that among the infections involving the GI-19 lineage, broilers were overrepresented. In infections involving GI-13 we found the opposite pattern: broilers were underrepresented and most GI-13 infected chickens were layers. Even though it is challenging to proclaim any definitive conclusion from this pattern, a possible scenario is that the major lineages differ in the ability to exploit different risk factors between the two farming types, such as differences in vaccination strategy. If the two lineages differ in their ability to sustain transmission between production types that would explain why the major lineages are not engaged in epidemic switching. Regardless of whether this scenario turns out to be correct, a better understanding of what factors allow the co-existence of different lineages as well as the impact of differences in risk factors between the different production types may be helpful to veterinarians.
Another intriguing difference between the two major IBV lineages is the turnover of novel variants within lineages. Both GI-13 and GI-19 include large clusters of very similar sequences. Close examination of these clusters in GI-13 reveals those to be vaccine-derived sequences. This stands to reason, since live attenuated vaccines remain unchanged and are usually administered at a large scale. Within GI-19 we found several clusters of sequences that could not be traced to known vaccines. Moreover, these clusters appeared in a succession of burst that typically lasted 1-3 years (Figure 1). We believe these to be consecutive epidemics that swept through the Dutch livestock populations. Alternatively, they may represent successive introductions and extinctions of field strains into livestock. Monitoring such sublineage turnover may turn out to be insightful in tracing the transmission routes between farms and aid in reducing the number of new infections.
Conclusion
Genetic typing of IBV sequences provides a high resolution for distinguishing the various lineages infecting chicken. The additional insights that are gained by studying lineage dynamics can provide valuable knowledge for veterinarians and poultry farmers.
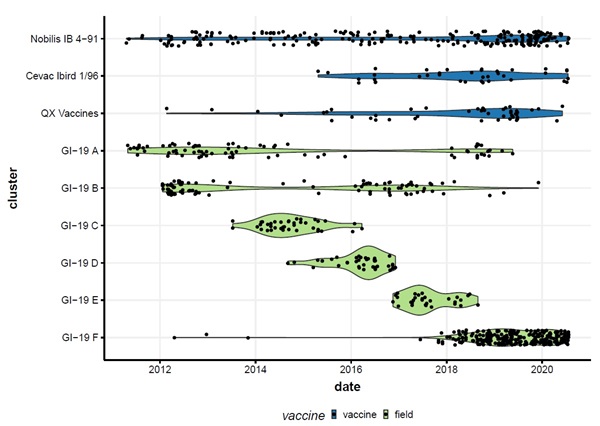 Figure 1. Succession of clusters of similar sequences in time. The clusters are overepresented in broiler chickens. The upper three clusters are known vaccines, with the similar Nobilis IB Primo QX and Poulvac IB pooled in “QX vaccines”. Note how the six non-vaccine clusters succeed each other in time.
This work was supported by the ZonMw under Grant 10430022010001.
Figure 1. Succession of clusters of similar sequences in time. The clusters are overepresented in broiler chickens. The upper three clusters are known vaccines, with the similar Nobilis IB Primo QX and Poulvac IB pooled in “QX vaccines”. Note how the six non-vaccine clusters succeed each other in time.
This work was supported by the ZonMw under Grant 10430022010001.
Reference: Vermeulen, C. J., Dijkman, R., de Wit, J. J., Bosch, B-J., Heesterbeek, J. A. P. & van Schaik, G. Genetic analysis of infectious bronchitis virus (IBV) in vaccinated poultry populations over a period of 10 years, Avian Pathol. (2023) https://doi.org/10.1080/03079457.2023.2177140